

 Реферат: Алкилирование енаминов, бета-дикетонов и енаминокетонов
Реферат: Алкилирование енаминов, бета-дикетонов и енаминокетонов
Енаминная система в щелочных средах может быть депротонирована и продукт введен в реакцию с алкилирующими агентами.
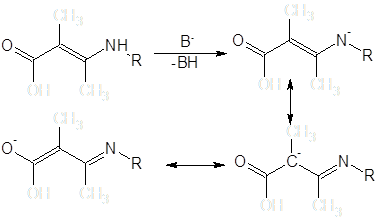
До последнего времени считалось, что преобладающими продуктами реакций алкилирования енаминокетонов являются N-замещенные производные [13]. Данные последующих исследований показывают, что алкилирование некоторых β-енаминокетонных систем в условиях межфазного переноса может быть селективно проведено и по атому углерода [14]:
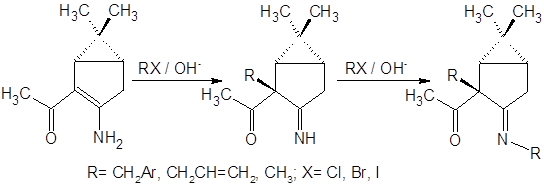
Приведенная последовательность превращений характерна, однако, только для алкилирования стерически нагруженными алкилгалогенидами. При использовании в качестве галогенида йодистого метила образуются все продукты вплоть до пентазамещенного. Исходя из данных настоящей работы, можно отметить, что направление алкилирования приведенных на рисунке соединений существенно зависит также от температуры реакции: при 35°С преобладает продукт С-алкилирования, при более низких температурах из смеси удается выделить N-алкилзамещенный продукт, доля С-замещения при этом невелика.
В работе [13] исследовано алкилирование 3-амино-5,5-диалкилциклогекс-2-ен-1-она для различных алкильных заместителей и проведен анализ факторов, необходимых для селективного направления алкилирования по тем или иным положениям изученного енаминона:
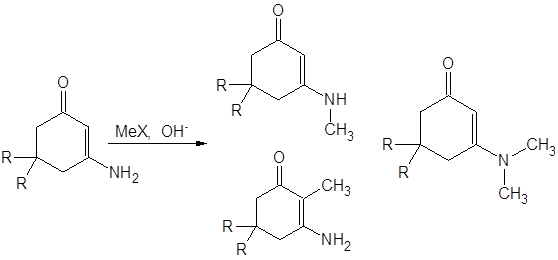 Зависимость хода реакции
алкилирования аналогичного [13] циклического енаминокетона от природы боковых
радикалов изучена авторами [3]:
Зависимость хода реакции
алкилирования аналогичного [13] циклического енаминокетона от природы боковых
радикалов изучена авторами [3]:
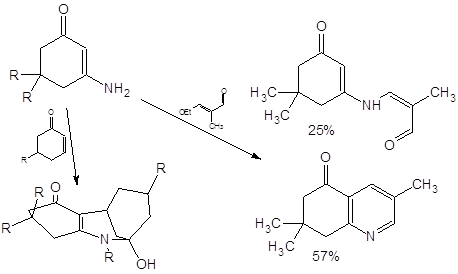
Среди известных реакций енаминонов внимания также заслуживает описанная в [15] реакция фотоарилирования:
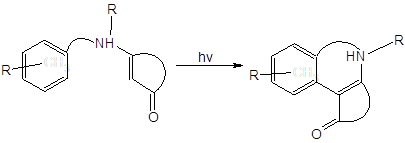
Авторы утверждают, что облучение енаминонов светом с длиной волны < 300 нм “can result in the formation of a variety of products… …photoreductions predominate”.
Из приведенных литературных данных можно сделать выводы об условиях, необходимых для получения высоких выходов С-алкилированных производных енаминов, b-дикетонов и енаминонов:
1. Необходим подбор основности среды. В низкоосновных средах мала концентрация активного аниона и реакция протекает медленно, в слишком высокоосновной среде происходит депротонирование атома азота и преобладающим становится продукт N-алкилирования.
2. Упомянутые выше требования к кинетике процесса алкилирования должны быть удовлетворены в максимальной степени.
3. С-алкилированный продукт, получающийся при повышенных температурах, является следствием термодинамического контроля реакции реакции алкилирования, при снижении температуры реакции возрастает доля продуктов кинетического контроля -- N- и O-алкилированных продуктов.
4. Асимметрическая индукция от имеющихся структурных фрагментов может обеспечивать отмеченное многими авторами стереоселективное протекание реакции алкилирования [2, 3, 5, 11, 12, 14].
В целом можно отметить, что несмотря на широкую известность описанных соединений, реакции алкилирования с их участием изучены пока недостаточно.
![]()
Экспериментальная часть.
Синтез (3-амино-6,6-диметилбицикло[3.1.0]гекс-2-ен-2-ил)метилкетона II:
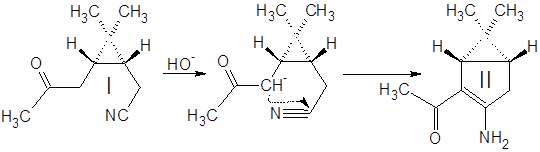
К 350 мл спиртового раствора KOH при перемешивании в течение 10 мин. добавили 100 г кетонитрила I. Смесь нагрели и кипятили с обратным холодильником 15 мин., охладили и разбавили водой в три раза. Провели экстракцию смеси метилтретбутиловым эфиром (300, 300, 150 мл), эфирную фазу экстрагировали 1М HCl (900, 500, 300 мл). Полученный водный раствор нейтрализовали 30% аммиаком и экстрагировали tBuOMe (200, 200, 100 мл). Эфирный раствор высушили MgSO4 безв и отогнали растворитель. Выход 79%.
Синтез (3-амино-2-бензил-6,6-диметилбицикло[3.1.0]гекс-2-ил)метилкетона III:
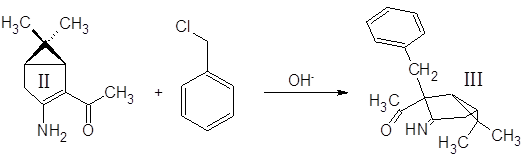
5 г енаминокетона II растворили в 30 мл бензола, в раствор добавили 15 мл 40% водного раствора NaOH и 0.5 г Bu4N+NO-3, перемешивали несколько минут и постепенно (3 мин.) добавили 10 мл бензилхлорида. Смесь интенсивно перемешивали 1.5 часа при 35-40˚С. Водную фазу отбросили, органическую экстрагировали 1М H2SO4 (15, 15, 15 мл). Экстракт нейтрализовали избытком 30% аммиака и экстрагировали метилтретбутиловым эфиром. Эфирный раствор высушили MgSO4 безв и отогнали растворитель. Выход 44%.
Синтез 1-(3-амино-6,6-диметилбицикло[3.1.0]гекс-2-ен-2-ил)этанола:
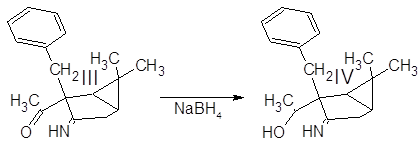
4 г алкилированого енаминокетона III растворили в 40 мл этанола. В раствор всыпали 0.6 г NaBH4 и перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь разбавили водой и экстрагировали эфиром. Эфирную фазу промыли водой для удаления спирта и вылили в водный раствор 4 г Cu(OAc)2·2H2O. Голубые кристаллы отфильтровали и высушили на воздухе.
Продукты исследовали методами хроматомасс-спектрометрии, ИК и ЯМР 13C и 1H. ЯМР спектры регистрировали на приборе Bruker DPX-500 (НИОХ СО РАН) в смеси CCl4/CDCl3; химсдвиги отсчитывали: в протонных спектрах – от сигнала остаточных протонов CDCl3 (7.250 м.д.), в спектрах 13С – от сигнала атома углерода CCl4 (96.10 м.д.). ИК спектры записывали на однолучевом спектрометре Bruker Vector 22 (256 усреднений с вычетом фона). Хроматомасс-спектрометрический анализ выполнен сотрудниками НИОХ.
Бензилхлорид и все растворители использовали свежеперегнанными. Точность отсчета температуры ±2°С, времени – ±2 мин. Тонкослойная хроматография выполнена на пластинках “Silufol”® (SiO2 на алюминиевой фольге).
Результаты и их обсуждение.
Из описанных выше реакций С-алкилирования наилучшим образом изучены реакции алкилирования b-дикетонов и енаминов. По енаминокетонам, несмотря на их широкое применение в синтезе, данных значительно меньше. Практическая потребность в проведении алкилирования и обнаруженная неоднозначность протекания этой реакции потребовали ее более детального изучения на конкретных соединениях. В качестве алкилирующего реагента был избран бензилхлорид, в условиях реакции не дающий продуктов полиалкилирования (метилирование в тех же условиях может быть четырех-пятикратным [14]).
Схема проведенных превращений такова:
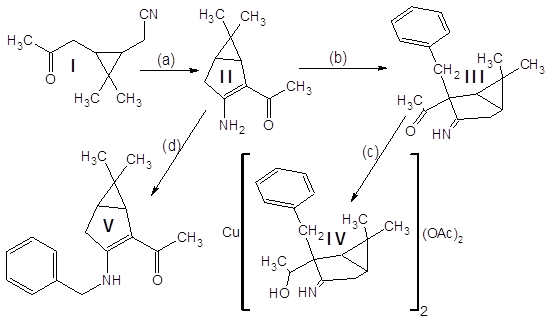
Кетонитрил I (исходное соединение, чистота ~90% (ГЖХ), предоставлен ЛТС НИОХ СО РАН) является производным природного терпена – 3-карена, выделяемого из соснового скипидара:
Результирующее соединение IV используется далее в синтезе оптически активных комплексных соединений, используемых в асимметрическом катализе.
Кетонитрил I представляет собой вязкую темную жидкость, растворимую в органических растворителях и нерастворимую в воде. Реакция (а) проходит гладко и с высоким выходом (~80%) дает продукт конденсации – (3-амино-6,6-диметилбицикло[3.1.0]гекс-2-ен-2-ил)метилкетон II.
Отдельного рассмотрения заслуживает процесс бензилирования соединения II. Эта реакция при комнатной температуре протекает крайне неоднозначно, с выходом целевого продукта менее 10% и образованием трудноразделимой смеси изомерных продуктов и продуктов полиалкилирования. Схема превращений и идентифицированные продукты представлены на следующем рисунке:
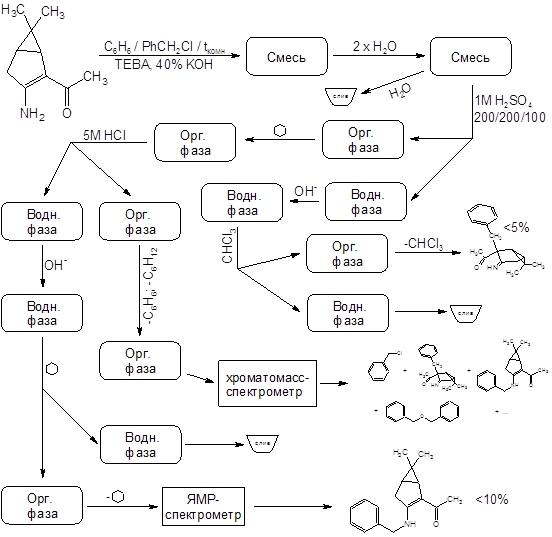
Показанные на схеме процессы обладают повторяемостью – по данным тонкослойной хроматографии в продуктах нескольких проведенных реакций набор компонентов один и тот же. Выходы выделенных веществ тоже примерно одинаковы. Спектры ядерного магнитного резонанса N-алкилированого продукта имеются в Приложениях. Его выход составляет 10%, но (!) – зимой. В тех же реакциях, осуществленных летом, продукт N-алкилирования зафиксирован не был.
Предположение о связи направления алкилирования с температурой реакционной смеси (а в методике указана комнатная температура) подтвердилось – проведение реакции при 35ºС дало в качестве преобладающего продукта (с выходом ~60%) целевой С-замещенный енаминокетон, алкилирование прошло так, как описано в “летней” методике. Попытки дальнейшего повышения температуры пока не предпринимались.
Полученный (3-амино-2-бензил-6,6-диметилбицикло[3.1.0]гекс-2-ил)метилкетон III был введен в реакцию с избытком боргидрида натрия для восстановлния карбонильной группы в спиртовую.
Точный состав и строение комплекса IV неизвестны – парамагнитный ион меди II препятствует получению спектров ЯМР, по данным ИК-спектра что-либо определенное сказать затруднительно. Сводка полученных характеристик соединений дана ниже:

Результаты проделанной работы можно суммировать следующим образом:
- синтезированы и охарактеризованы спектрально и хроматографически некоторые производные природного терпена 3-карена.
